
28 DE JUNIO. DIA MUNDIAL DE LA FENILCETONURIA O PKU
 Cada 28 de junio se celebra el Día Mundial de la Fenilcetonuria, con la finalidad de visibilizar esta enfermedad rara que tiene una prevalencia de 1 por cada 12.000 nacimientos. La creación de esta efeméride en el año 2013 fue por iniciativa de la Sociedad Europea de Fenilcetonuria, en Amberes (Bélgica). Doña Águeda Barcina. Enfermera. Socia ACYLEU
Cada 28 de junio se celebra el Día Mundial de la Fenilcetonuria, con la finalidad de visibilizar esta enfermedad rara que tiene una prevalencia de 1 por cada 12.000 nacimientos. La creación de esta efeméride en el año 2013 fue por iniciativa de la Sociedad Europea de Fenilcetonuria, en Amberes (Bélgica). Doña Águeda Barcina. Enfermera. Socia ACYLEU
¿Qué es la Fenilcetonuria o PKU?
Es el trastorno hereditario más frecuente del metabolismo de los aminoácidos. Se caracteriza por un incremento en la concentración sanguínea y tisular del aminoácido esencial fenilalanina (Phe) como resultado de la deficiencia del enzima fenilalanina hidroxilasa, que cataliza la conversión de Phe en tirosina (Tyr). Esta deficiencia presenta un patrón de herencia autosómico recesivo y está causada por variantes patogénicas en el gen PAH que codifica dicho enzima
Sintomatología asociada:
Los recién nacidos que tienen fenilcetonuria al principio no presentan ningún síntoma. Sin embargo, sin tratamiento, los bebés suelen manifestar signos de fenilcetonuria en pocos meses.
-
-
- Olor a humedad en el aliento.
- Problemas neurológicos (convulsiones).
- Erupciones cutáneas (eczema).
- Retrasos en el desarrollo.
- Discapacidad intelectual.
- Ojos azules y piel clara, debido a que la fenilalanina no puede transformarse en melanina.
- Microcefalia
- Discapacidad intelectual.
- Hiperactividad
- Retraso en el desarrollo.
- Problemas de comportamiento, emocionales y sociales
- Trastornos psiquiátricos.
-
Factores de riesgo:
- Que ambos padres tengan un gen defectuoso que provoca la fenilcetonuria.
- Ser de cierta ascendencia étnica: menos frecuente en los afroamericanos que en otros grupos étnicos.
Tipos:
La gravedad de la fenilcetonuria depende del tipo.
- Fenilcetonuria clásica: Es la forma más grave. La enzima necesaria para convertir la fenilalanina es inexistente o está disminuida en extremo, lo que produce niveles elevados de fenilalanina y un daño cerebral grave.
- Formas menos graves de fenilcetonuria: En las formas leves o moderadas, la enzima conserva alguna función, por lo tanto, los niveles de fenilalanina no son demasiado elevados, lo que produce un menor riesgo de daño cerebral significativo.
¿Cómo se hereda?
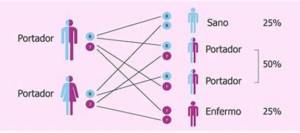
Tanto la madre como el padre deben tener el gen defectuoso y transmitirlo para que su hijo padezca fenilcetonuria. Este patrón de herencia se denomina «autosómico recesivo». Si solo uno de los padres tiene el gen de fenilcetonuria, no hay riesgo de transmitirle la enfermedad al hijo, pero es posible que este último sea portador.
Es más frecuente que dos padres que son portadores de este trastorno sin saberlo les transmitan la fenilcetonuria a sus hijos.
Diagnóstico en Castilla y León:
En el SACYL existe el Programa de Detección Precoz de Enfermedades Congénitas en Castilla y León.
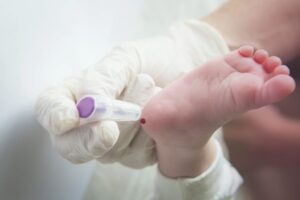
Es un programa de cribado poblacional cuya finalidad es la detección precoz de alteraciones metabólicas y genéticas mediante la determinación de diversos parámetros en muestras de sangre procedente del talón del recién nacido tomadas entre las 48 y 72 horas de vida, con el objetivo de disminuir la incidencia de deficiencias físicas y psíquicas.
Tratamiento:
No hay cura para la fenilcetonuria (PKU), pero el tratamiento puede prevenir las discapacidades intelectuales y otros problemas de salud.

- Una dieta de por vida muy limitada en proteínas.
- Leche y queso
- Huevos
- Nueces
- Soja
- Pollo, carne de res o cerdo
- Pescado
- Cerveza
- Tomar leche maternizada para la fenilcetonuria.
- Es especialmente importante que una mujer embarazada con fenilcetonuria siga estrictamente la dieta baja en fenilalanina a lo largo de su embarazo para garantizar el desarrollo saludable de su bebé.
- Evitar el edulcorante Aspartamo.
Bibliografía:
- ORDEN PRE/1863/2022, de 16 de diciembre. Carta de servicios del programa de detección precoz de enfermedades congénitas en Castilla y León.
- American College of Medical Genetics and Genomics Therapeutic Committee: Diagnosis and management guidelines for phenylalanine hydroxylase deficiency (2013)
- (2000, actualizado en 2006). Report of the NIH consensus development conference on phenylketonuria (PKU): Screening and management. Obtenido el 24 de junio de 2024 de https://www.nichd.nih.gov/publications/pubs/pku/index.