11 de abril – DIA MUNDIAL DEL PARKINSON

«En abril de 1997 Parkinson’s Europe impulsó el Día Mundial del Parkinson cuando se llamaba Asociación Europea de la Enfermedad de Parkinson o EPDA. El evento fue copatrocinado por la Organización Mundial de la Salud. Esta fecha coincide con el aniversario del nacimiento de James Parkinson, un neurólogo británico que en 1817 descubrió lo que en aquel tiempo denominó parálisis agitante y que hoy conocemos como enfermedad de Parkinson». Doña Águeda Barcina. Enfermera. Socia ACYLEU

¿Qué es el Parkinson?
La enfermedad de Parkinson es una condición degenerativa, progresiva y crónica del sistema nervioso que se caracteriza por causar severos daños neurológicos, generando alteraciones en el control y coordinación de los movimientos del cuerpo, así como rigidez muscular.
El daño aparece cuando las células del sistema encargadas de producir dopamina, una hormona que regula el movimiento, detienen su producción y esto termina por desencadenar la enfermedad.
Los síntomas aparecen de forma progresiva, afectando algunas zonas del cuerpo como las manos, los brazos, las piernas y la cara. Luego se extiende a todo el cuerpo, causando rigidez motora, temblores, problemas de equilibrio y coordinación.
¿Sabías que…..?
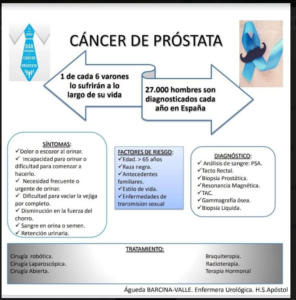
- Según la Organización Mundial de la Salud, la enfermedad de Parkinson afecta a 1 de cada 100 personas mayores de 60 años.
- La Asociación Europea de la Enfermedad de Parkinson (EPDA) estima que más de 6 millones de personas en el mundo padecen de Parkinson.
- Unas 160.000 personas padecen Parkinson en España. Se estima que la cifra se acerca más a los 300.000 afectados y que para el año 2030 habrá unos 12 millones de pacientes con Parkinson.
- Cada año se diagnostican unos 10.000 nuevos casos de la enfermedad de Parkinson en España.
- Entre el 20 y 40% de los pacientes presenta depresión, como un síntoma precoz del Parkinson.
- Es la segunda enfermedad neurodegenerativa después del Alzheimer
- El 70% de las personas diagnosticadas de Parkinson tienen más de 65 años,
- El 15% son menores de 50 años.
- Los pacientes tardan una media de entre 1 y 3 años en obtener un diagnóstico
- Entre el 20 y el 25% de los pacientes afectados por la enfermedad de Parkinson requieren un ingreso hospitalario anual.
Sintomatología
La enfermedad de Parkinson causa síntomas motores, como:

- lentitud de movimientos
- temblor
- movimientos involuntarios
- rigidez
- dificultad para andar
- pérdida del equilibrio.
Síntomas no motores:
- deterioro cognitivo
- trastornos mentales
- demencia
- trastornos del sueño
- dolor
- alteraciones sensoriales
Aunque la enfermedad de Parkinson es el trastorno del movimiento más común, existen otros trastornos del movimiento, como la atrofia multisistémica, la parálisis supranuclear progresiva, la corea, la ataxia y la distonía. Algunos trastornos del movimiento presentan síntomas similares a los de la enfermedad de Parkinson, como el temblor, la lentitud de movimientos y la rigidez. Todos los trastornos del movimiento comportan los mismos retos que la enfermedad de Parkinson en lo que respecta a las brechas en materia de diagnóstico y tratamiento y al acceso a los medicamentos, especialmente en los países de ingreso bajo y mediano.
El tulipán, el símbolo del Parkinson
 En el año 2005 se eligió un tulipán rojo como símbolo de la enfermedad de Parkinson en una reunión sobre la enfermedad celebrada en Luxemburgo.
En el año 2005 se eligió un tulipán rojo como símbolo de la enfermedad de Parkinson en una reunión sobre la enfermedad celebrada en Luxemburgo.
La historia de este símbolo se retrotrae al año 1980 cuando el horticultor holandés JWS Van der Wereld, que padecía Parkinson, desarrolló una variante roja y blanca del tulipán. Lo llamó tulipán del Dr. James Parkinson en honor al médico inglés que escribió por primera vez sobre esta afección en su publicación de 1817, «El ensayo sobre la parálisis temblorosa».
A partir de 2005, muchas organizaciones han adaptado el símbolo del tulipán como imagen. Y es un buen ejemplo para explicar la enfermedad, ya que un tulipán descuidado empezará a doblarse y marchitarse pronto, pero si se le cuida durará más tiempo en buen estado. Al igual que el tulipán, un paciente de Parkinson que se cuide, se tome su medicación y haga deporte, podrá conservar su salud más tiempo.
Terapias y nuevos avances contra la enfermedad de Parkinson
Aunque no hay cura para la enfermedad de Parkinson, los tratamientos, en particular los medicamentos, la cirugía y la rehabilitación, pueden reducir los síntomas.
- La levodopa/carbidopa, un medicamento combinado que aumenta la cantidad de dopamina en el cerebro, es el medicamento más común para la enfermedad de Parkinson. Los médicos pueden usar otros medicamentos para reducir el movimiento muscular involuntario, como los anticolinérgicos.
- La estimulación cerebral profunda y otros tratamientos pueden ayudar con los temblores y reducir la necesidad de medicamentos.
- La rehabilitación, incluida la fisioterapia, puede ofrecer alivio para la enfermedad de Parkinson y en el caso de otros trastornos neurológicos degenerativos. Las opciones de tratamiento incluyen:
-
-
- entrenamiento de fuerza.
- ejercicios de movilidad y equilibrio
- hidroterapia.
Estos tratamientos pueden ayudar a mejorar el funcionamiento y la calidad de vida de las personas con la enfermedad de Parkinson, así como reducir la presión sobre quienes atienden a las personas con esta enfermedad.
También, cabe destacar un estudio que se está realizando en España, donde las personas son sometidas a una terapia que estimula el movimiento, utilizando la realidad virtual. A través del uso de un caso y un video juego, se puede ayudar al paciente a trabajar el movimiento, controlando y monitoreando los impulsos eléctricos del cerebro.
















 ¡
¡



