¿Por qué se celebra el Día Mundial del Vitíligo el 25 de junio?
La elección de la fecha para el Día Mundial del Vitíligo el 25 de junio es un homenaje a Michael Jackson, que sufrió esta enfermedad desde principios de la década de los ochenta hasta su muerte, precisamente el 25 de junio de 2009. Se eligió el aniversario de su fallecimiento para conmemorar esta enfermedad y crear conciencia.
¿Qué es?
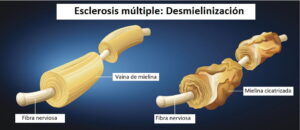
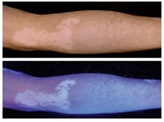
Es una enfermedad autoinmune y progresiva de la piel, caracterizada por la aparición de manchas o lesiones acrómicas de diversos tamaños denominadas máculas, de color blanco nacarado y con una superficie lisa.
Es ocasionado por la destrucción de los melanocitos (células responsables de la pigmentación de la piel), disminuyendo la producción de melanina en varias partes del cuerpo, tales como contorno de los ojos, fosas nasales, cara, codos, tobillos, axilas, rodillas, manos y pies.
Si la lesión aparece en áreas pilosas (cuero cabelludo, pestañas, cejas y genitales) afectará el color del vello, conocido como poliosis.
Este trastorno se vincula a factores tales como predisposición genética, estrés y se ha asociado a otras enfermedades (diabetes, Enfermedad de Adisson, anemia perniciosa y trastornos tiroideos).
Es una enfermedad que afecta al 2% de la población mundial, siendo más frecuente en aquellas razas que tienen mayor cantidad de pigmentación en la piel.
Hay una mayor prevalencia en mujeres y la edad de aparición se encuentra comprendida entre los 10 y 30 años, aunque puede aparecer en cualquier momento de la vida.
Tipos de vitíligo
- Aparecen máculas aisladas y reducidas en tamaño y número en cualquier localización.
- Las máculas son unilaterales y suelen seguir una distribución determinada.
- Es el más común y se caracteriza por múltiples máculas hipopigmentadas dispersas por toda la superficie corporal, de disposición simétrica.
- Afecta partes distales y a la región facial. La forma universal es aquella en la que quedan pocas áreas corporales pigmentadas.
Vitíligo: una anomalía estética no aceptada
Por ser una enfermedad visible, que afecta estéticamente al paciente, puede ocasionar rechazo o poca aceptación en su entorno social y familiar. Esto puede llegar a generar baja autoestima, ideación suicida (en algunos casos), estrés, irritabilidad, agresividad, aislamiento social, depresión y ansiedad.
Resulta pertinente buscar apoyo profesional especializado (dermatólogo, psicólogo), a fin de efectuar un adecuado seguimiento y control del paciente, aunado al apoyo familiar para afrontar esta enfermedad.

Es de vital importancia concienciar a la población mundial sobre el vitíligo, para normalizar esta condición o trastorno en la Sociedad y evitar la discriminación y rechazo hacia las personas que la padecen.
Diagnóstico, tratamiento y recomendaciones
El diagnóstico del vitíligo es efectuado por un dermatólogo, mediante una evaluación clínica en la piel del paciente, utilizando una lámpara de hendidura o de Wood.
En algunos casos, puede ser necesaria una biopsia de piel para verificar otras posibles causas de la pérdida de pigmentación. Así como analítica de sangre con determinación de hormonas tiroideas y de vitamina B12.
Opciones de tratamiento dependiendo del grado y ubicación de las lesiones, así como la edad del paciente.
- Corticoides tópicos.
- Fototerapia con radiación Ultravioleta y UVB de banda estrecha.
- Antioxidantes orales.
- Inhibidores de calcineurina.
- Injerto de piel.
- Trasplante de suspensión celular.
En estos pacientes es fundamental el cuidado de la piel:
- Proteger la piel del sol, así como de fuentes artificiales de luz ultravioleta.
- Utilizar protector solar con un elevado factor de protección y resistente al agua.
- Utilizar prendas de vestir que protejan la piel del sol.
- Evitar el uso de camas bronceadoras y lámparas solares.
- Evitar la colocación de tatuajes, especialmente en la zona de las lesiones.
- Consumir vitaminas (Ginko Biloba, vitamina C, vitamina B-12, ácido fólico y ácido alfa-lipoico).
Puede encontrar más información sobre el vitiligo en:
Águeda Barcina Valle. Socia ACYLEU


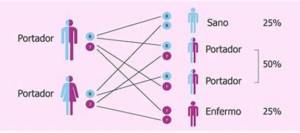
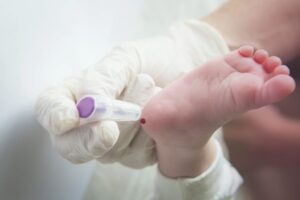
 Cada 28 de junio se celebra el Día Mundial de la Fenilcetonuria, con la finalidad de visibilizar esta enfermedad rara que tiene una prevalencia de 1 por cada 12.000 nacimientos. La creación de esta efeméride en el año 2013 fue por iniciativa de la Sociedad Europea de Fenilcetonuria, en Amberes (Bélgica). Doña Águeda Barcina. Enfermera. Socia ACYLEU
Cada 28 de junio se celebra el Día Mundial de la Fenilcetonuria, con la finalidad de visibilizar esta enfermedad rara que tiene una prevalencia de 1 por cada 12.000 nacimientos. La creación de esta efeméride en el año 2013 fue por iniciativa de la Sociedad Europea de Fenilcetonuria, en Amberes (Bélgica). Doña Águeda Barcina. Enfermera. Socia ACYLEU